
Введение
Компьютерные эксперименты играют очень важную роль в науке. В реальном физическом эксперименте проводится измерение характеристик изучаемой системы и получаются результаты, выраженные в числовой форме. В теории модель системы строится обычно в форме набора математических уравнений. Затем проверяется способность модели описывать поведение системы на нескольких выбранных вариантах реализации модели, достаточно простых, чтобы позволить найти решение уравнений. Во многих случаях это подразумевает значительное количество упрощений, чтобы устранить все сложности, связанные с проблемами реальной среды, и сделать задачу решаемой.
Что такое молекулярная динамика
Молекулярной динамикой (MД) называется метод компьютерного моделирования, где эволюция во времени ансамбля взаимодействующих атомов определяется интегрированием уравнений их движения. Рассмотрим кратко основы метода МД моделирования свойств материалов. Если поведение атомных частиц подчиняется законам классической или квантовой механики, то их движение описывается динамикой Гамильтона . Поэтому рассматриваемая атомная система (ансамбль) в любой фиксированный момент времени характеризуется совокупностью обобщённых координат и обобщённых импульсов,
 где N- число частиц в рассматриваемом ансамбле.
где N- число частиц в рассматриваемом ансамбле.Микроскопическое состояние системы определяется численным значением переменных
 . Различные свойства системы описываются динамическими функциями b(p, q), зависящими от обобщённых координат и импульсов. Эти функции принимают определенные значения в каждом микроскопическом состоянии (p, q) системы.
. Различные свойства системы описываются динамическими функциями b(p, q), зависящими от обобщённых координат и импульсов. Эти функции принимают определенные значения в каждом микроскопическом состоянии (p, q) системы. В динамики Гамильтона движение полностью определено, если для системы задан гамильтониан Н(p, q). Эта функция полностью характеризует динамическую природу системы. С физической точки зрения, в большинстве случаев (но не всегда) гамильтониан представляет полную энергию системы. При эволюции Гамильтоновой динамической системы изображающая точка двигается в фазовом пространстве (p, q) по определённой траектории. Эту траекторию можно представить как совокупность 2N функций времени
 , которые определяются из решения уравнения Гамильтона(1):
, которые определяются из решения уравнения Гамильтона(1): 
Такая система 2N дифференциальных уравнений позволяет определить единственным образом значения переменных (p_i, q_i) для любого момента времени по их начальным значениям
 . Для ансамбля бесструктурных частиц, взаимодействующих через эффективный потенциал, зависящий только от взаимного расположения частиц, обобщенные координаты, импульсы и гамильтониан определяются следующими соотношениями:
. Для ансамбля бесструктурных частиц, взаимодействующих через эффективный потенциал, зависящий только от взаимного расположения частиц, обобщенные координаты, импульсы и гамильтониан определяются следующими соотношениями: 
Соответственно, система уравнений (1) приводится к виду (2)

 как систему уравнений (2) можно записать в виде(3):
как систему уравнений (2) можно записать в виде(3):
Системы уравнений (3) или (4) представляют классические уравнения движения Ньютона. Возможность применения классических уравнений движения для описания состояния микрочастиц (атомов или молекул) определяется условием lamba << a, где lamba - длина волны де-Бройля для рассматриваемой частицы, a-характерное расстояние между частицами. Длина волны де-Бройля для частицы массы
 определяется соотношением(5):
определяется соотношением(5):
Учитывая численные значения фундаментальных констант
 выражение (5) можно привести к виду
выражение (5) можно привести к виду

Выражение (5) получено при значении температуры Т=300 К. В таблице 1 приведены рассчитанные величины отношения для некоторых металлов и газов. Для металлов величина отождествлялась с межатомным расстоянием между ближайшими соседями в кристаллической решетке при нормальных условиях. Для газов величина отождествлялась с параметром s в межчастичном потенциале Леннарда-Джонса. Численные значения параметров s взяты из литературных источников.
Таблица 1. Численные значения отношения длины волны де-Бройля к характерному межатомному расстоянию
|
Вещество |
|
Молекулярный |
 |
|
C
(алмаз) |
1.544 |
12.011 |
0.187 |
|
Al |
2.863 |
26.981 |
6.72. 10-2 |
|
Cu |
2.556 |
63.546 |
4.91. 10-2 |
|
W |
2.741 |
183.85 |
2.69. 10-2 |
|
Ar |
3.418 |
39.948 |
4.63. 10-2 |
|
N2 |
3.749 |
28.013 |
5.034 10-2 |
|
H2 |
2.968 |
2.016 |
0.237 |
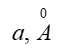
Как видно из приведенных в таблице 1 результатов, условие
lamba/a<< 1 выполняется для всех металлов. При нормальных и повышенных температурах это условие выполняется так же и для многих газов.
Траектории и скорости частиц, рассчитанные при решении системы уравнений (1.3) или (1.4), позволяют получить информацию о термодинамических и кинетических свойствах вещества. Переход от микроскопических характеристик атомных ансамблей, которые получаются в результате МД моделирования, к макроскопическим свойствам материалов осуществляется на основе методов статистической механики.
Области применения молекулярной динамики
Область применения методов МД моделирования очень широка. Ниже приведены только некоторые приложения и, конечно, представленный список нельзя рассматривать как исчерпывающий:
Жидкости. Исследование структуры различных жидкостей является важным приложением МД метода. Разработанные современные модели межатомных взаимодействий позволяют изучать новые системы, элементарные и многокомпонентные. С помощью неравновесных методов исследуются явления переноса, например, вязкость и теплопроводность.
Дефекты. Другое традиционное приложение МД моделирования – это изучение влияния дефектов на механические свойства кристаллов, что представляет большой практический интерес и по-прежнему остается актуальной темой физики твёрдого тела. В настоящее время интерес сместился от точечных дефектов (вакансии, междоузлия) к линейным (дислокации) и плоским (границы зерна, дефекты упаковки).
Разрушение. При определённом уровне механического воздействия твёрдые тела разрушаются. Процесс разрушения может происходить различными способами и с различной скоростью, которая зависит от многих параметров. Технологическая важность понимания этого процесса очевидна и МД моделирование позволяет понять микроскопические механизмы процесса разрушения твёрдых тел.
Поверхности. Физика поверхностей в настоящее время интенсивно развивается. Появились новые уникальные экспериментальные инструменты с микроскопическим разрешением (сканирующая микроскопия на основе туннельного эффекта, электронная микроскопия высокого разрешения, несколько методов на основе атомного рассеивания). МД моделирование здесь играет большую роль в понимании таких явлений, как поверхностные реконструкции, поверхностное плавление, образование граней, поверхностная диффузия.
Кластеры. Кластеры – это группы атомов, от нескольких штук до нескольких тысяч атомов. Атомные или молекулярные кластеры по своим физико-химическим свойствам отличаются от макроскопического образца материала. Например, характеристики кластеров могут значительно отличаться от свойств твердого вещества вследствие их малого размера, большого влияния поверхности и анизотропии. Металлические кластеры чрезвычайно важны с технологической точки зрения, благодаря их роли как катализаторов в важных химических реакциях (например, в каталитических ловушках выхлопных газов автомобилей).
Ударные волны. Методы МД моделирования широко применяются для изучения структуры ударных волн в конденсированных средах. Пространственный и временной масштаб физико-химических процессов, происходящих в ударном скачке, идеально подходит для изучения этих процессов методом МД моделирования.
Электронные свойства и динамика. Развитие метода Кара (Car)-Паринелло (Parrinello), когда силы, действующие на атомы, вычисляются путем решения задачи электронной структуры атомного ансамбля вместо использования межатомного потенциала, позволяет изучать электронные свойства материалов, полностью включая их динамику.
Ограничения метода МД моделирования
Молекулярная динамика очень мощный метод компьютерного моделирования, но, конечно, он имеет свои ограничения.
Ограничения, обусловленные выбором межатомного потенциала
В молекулярной динамике атомы непрерывно взаимодействуют друг с другом. Эти взаимодействия порождают силы, которые действуют на атомы, и атомы приобретают ускорения под действием этих мгновенных сил. При движении атомов изменяются их относительные положения, а также меняются значения силы, действующей на атомы. Следовательно, существенным элементом, заключающим в себе физику, являются силы. Соответственно, МД моделирование является реалистичным, то есть, моделирует поведение реальной системы в той степени, в какой межатомные силы эквивалентны тем, которые испытали бы реальные атомы (или, более точно, ядра) в такой же конфигурации.
Как описано ранее, силы обычно получаются как градиент функции потенциальной энергии в зависимости от положения частиц. Следовательно, реализм МД моделирования зависит от свойств потенциала, выбранного для воспроизведения поведения вещества в условиях, для которых выполняется моделирование.
Ограничения, обусловленные временем интегрирования и размерами ансамбля
В настоящее время с использованием суперкомпьютеров МД моделирование может проводиться на ансамблях, включающих десятки миллиардов атомов. Время моделирования, в зависимости от размера ансамбля, может лежать в пределах от нескольких пикосекунд до сотен наносекунд. Хотя эти значения являются допустимыми, можно столкнуться с условиями, при которых ограничения на время моделирования или ограничения на размер ансамбля становятся значимыми. Существует критерий, который определяет требуемое время моделирования. Время моделирования должно быть много больше времени релаксации интересующих нас физических величин. Очевидно, что различные свойства материалов имеют разные времена релаксации. В частности, физические процессы имеют тенденцию становиться медленными и малоподвижными вблизи фазовых переходов, и иногда можно столкнуться с ситуацией, когда время релаксации физического параметра на порядок большее времени выполнения моделирования.
Ограниченный размер, используемого для моделирования ансамбля, также может составить проблему. В этом случае необходимо сравнивать размер MД ячейки с корреляционными длинами интересующих пространственных корреляционных функций. Корреляционные длины могут также увеличиваться или даже расходиться вблизи фазовых переходов, и, если они становятся сопоставимыми с длиной МД ячейки (бокса), результаты становятся ненадежными.
Как описано ранее, силы обычно получаются как градиент функции потенциальной энергии в зависимости от положения частиц. Следовательно, реализм МД моделирования зависит от свойств потенциала, выбранного для воспроизведения поведения вещества в условиях, для которых выполняется моделирование.
Ограничения, обусловленные временем интегрирования и размерами ансамбля
В настоящее время с использованием суперкомпьютеров МД моделирование может проводиться на ансамблях, включающих десятки миллиардов атомов. Время моделирования, в зависимости от размера ансамбля, может лежать в пределах от нескольких пикосекунд до сотен наносекунд. Хотя эти значения являются допустимыми, можно столкнуться с условиями, при которых ограничения на время моделирования или ограничения на размер ансамбля становятся значимыми. Существует критерий, который определяет требуемое время моделирования. Время моделирования должно быть много больше времени релаксации интересующих нас физических величин. Очевидно, что различные свойства материалов имеют разные времена релаксации. В частности, физические процессы имеют тенденцию становиться медленными и малоподвижными вблизи фазовых переходов, и иногда можно столкнуться с ситуацией, когда время релаксации физического параметра на порядок большее времени выполнения моделирования.
Ограниченный размер, используемого для моделирования ансамбля, также может составить проблему. В этом случае необходимо сравнивать размер MД ячейки с корреляционными длинами интересующих пространственных корреляционных функций. Корреляционные длины могут также увеличиваться или даже расходиться вблизи фазовых переходов, и, если они становятся сопоставимыми с длиной МД ячейки (бокса), результаты становятся ненадежными.
Молекулярная динамика - теория или эксперимент?
Компьютерное моделирование выглядит иногда как теория, иногда как эксперимент. С одной стороны, мы имеем дело с моделями, а не с "реальным объектом", и это дает основание классифицировать компьютерное моделирование как теоретический метод. С другой стороны, процедура подтверждения справедливости модели при компьютерном моделировании очень напоминает эксперимент: мы запускаем расчеты и затем анализируем результаты в значительной степени тем же самым способом, как это делают физики-экспериментаторы.